



O. Marotta, F. Catapano, V. Marotta
UOC di Otorinolaringoiatria, Azienda Ospedaliera di Rilievo Nazionale e di Alta Specializzazione di Caserta
ABSTRACT
La sindrome di Klippel-Trenaunay (KTS) è una rara patologia congenita caratterizzata da malformazioni dei capillari (port-wine stains o emangiomi piatti), ipertrofia del tessuto molle e dell’osso, ampie vene varicose. Le malformazioni vascolari intracraniche sono molto rare. Gli Autori riportano il caso di una bambina di 4 anni con KTS, affetta da ipoacusia neurosensoriale profonda in orecchio sinistro e grave in quello destro, che fu sottoposta ad impianto cocleare presso la nostra struttura; nel nostro caso l’osso temporale era sede di formazioni angiomatose ed una di queste interessava la porzione verticale del n. facciale.
INTRODUZIONE
La sindrome di Klippel-Trenaunay (KTS) fu descritta originariamente da Maurice Klippel e Paul Trenaunay nel 1900 (1). Essa è una rara sindrome angio-osteoipertrofica caratterizzata dalla triade sintomatologica: malformazioni dei capillari (port-wine stains o emangiomi piatti), ipertrofia del tessuto molle e dell’osso, ampie vene varicose (2). Di solito coinvolge un arto inferiore, raramente coinvolge più arti, il tronco, il volto o il corpo intero. Sono molto rari i casi di angiomi intracranici associati con KTS. Viene definita Sindrome di Klippel-TrenaunayWeber se sono presenti anche fistole artero-venose (3). Sono molto rari i casi di angiomi intracranici associati con KTS (4, 5, 6). L’incidenza è bassa (1/27500 nascite). L’eziologia è sconosciuta. La malattia è congenita ed è dovuta ad una disfunzione dei meccanismi di angiogenesi e vasculogenesi nei siti interessati (7). Solo pochi casi in letteratura sono riportati di KTS associata a una perdita uditiva. In un lavoro del 2004 di Suver (8) si fa cenno a due casi: uno con ipoacusia neurosensoriale e uno con ipoacusia mista in una adolescente con fistole artero-venose causanti un iperaccrescimento dell’osso che si estendeva dalla coclea fin dentro la cavità dell’orecchio medio. In entrambi i casi le tipiche malformazione della KTS riguardavano il volto; ciò suggeriva, secondo l’autore, la necessità di eseguire una valutazione audiologica nei pazienti affetti da KTS interessante la testa. Gli Autori non hanno trovato in letteratura dati concernenti l’applicazione di impianto cocleare in pazienti affetti da KTS.
CASO CLINICO
Noi riportiamo il caso di una paziente, C. G. di anni 3, venuta alla nostra osservazione per ipoacusia con ritardo del linguaggio. All’anamnesi, i genitori riferivano che, in età neonatale, fu diagnosticata una KTS ed, in seguito a valutazione audiologica, le fu riscontrata sordità bilaterale, per cui, all’età di 5 mesi, la bimba fu sottoposta a protesizzazione bilateralmente e avviata a riabilitazione logopedica. A tre anni subì intervento di adenoidectomia e drenaggio transtimpanico bilaterale. Nonostante la terapia logopedica, a 4 anni, la paziente presentava un evidente ritardo del linguaggio. Per tale motivo viene alla nostra osservazione e sottoposta a valutazione per impianto cocleare. All’esame obiettivo, la paziente presentava un voluminoso angioma sul volto e sull’orecchio, collo e spalla sinistra; sclere di colore bluastro (fig. 1 e 2) Alla orofaringoscopia era evidente una asimmetria tonsillare con la tonsilla sinistra più voluminosa della destra. Le membrane timpaniche erano integre con iperemia della sinistra. Fu sottoposta, come tutti i pazienti candidati ad applicazione di I.C. presso la nostra U.O., ad un protocollo di valutazione preoperatorio che comprende: colloquio, anamnesi e visita ORL, valutazione audiologica, valutazione logopedica, valutazione psicologica, valutazione neurologica – neurochirurgica, valutazione neuropsichiatrica, esami di laboratorio, valutazione cardiologica, radiografia del torace, T.C. rocche petrose, R.M. encefalo, visita anestesiologica. La valutazione audiologica rivelò anacusia sx ed ipoacusia neurosensoriale grave-profonda a dx (fig. 3). L’esame dei potenziali evocati uditivi (ABR) evidenziò assenza di onde significative in orecchio sinistro; la quinta onda era presente fino a 90dB in orecchio destro (fig. 4). Alla valutazione logopedica presentava un ritardo del linguaggio. La tomografia computerizzata (TC) mostrò una coclea normoconformata bilateralmente e presenza di tessuto simil infiammatorio mastoideo (fig. 5). La risonanza magnetica nucleare (RM), oltre alla normale conformazione delle strutture labirintiche, evidenziò una emiipertrofia del tessuto cerebrale, in particolare della sostanza bianca (fig. 6).
INTERVENTO
Gli Autori decisero di sottoporre la ragazza ad intervento chirurgico di impianto cocleare a sinistra. La valutazione anestesiologica fu molto accurata e mirata a evitare soprattutto eventuali complicanze emodinamiche; la paziente fu valutata ASA II. Fu effettuata una mastoidectomia “a minima”. Si evidenziò la presenza di tessuto gelatinoso rossastro che occupava pressoché totalmente le cellette mastoidee e che, estendendosi attraverso l’antro, andava ad occludere l’epitimpano ed inglobare parzialmente la catena ossiculare. Nell’eseguire la timpanotomia posteriore fu riscontrata la presenza di un grosso vaso che avvolgeva per un tratto di 5 mm la porzione verticale del nervo facciale (fig. 7). Si procedette a coagulazione con pinza bipolare della formazione angiomatosa e alla sua rimozione, tenendo cura di non provocare lesioni del nervo; fu così possibile completare la timpanotomia posteriore (fig. 8). Fu posizionato il ricevitore stimolatore (RS) e, dopo aver effettuato la cocleostomia antero-inferiormente alla finestra rotonda, inserito il multielettrodo (ME) nella coclea. Quindi, si praticarono le prove di telemetria e il riflesso stapediale, che diedero esito positivo. Il decorso postoperatorio fu regolare. Il giorno successivo all’intervento fu eseguita RX della rocca petrosa, in proiezione di Stenvers modificata, che confermò il corretto posizionamento del ME (fig. 9). Quindi, la paziente fu dimessa. Circa un mese dopo l’intervento fu attivato l’IC. Seguirono controlli audiometrici periodici e trattamento logopedico.
DISCUSSIONE
La KTS è una sindrome raramente associata a disturbi uditivi (1, 2, 8). La presenza di coinvolgimento del distretto cervico facciale, evidente in casi come il nostro con una estesa angiomatosi cutanea e macrocrania, richiede sempre un approfondimento audiologico (8). In caso di applicazione di IC in un paziente affetto da KTS è necessario praticare un accurato studio neuroradiologico. Infatti, pur se rare le lesioni neurovascolari nella KTS, esso può evitare eventuali gravi complicazioni legate alla presenza di malformazioni artero-venose cerebrali (4, 5, 6). Risulta cruciale il ruolo della TC e RM, alfine di evidenziare eventuali malformazioni vascolari interessanti la rocca petrosa. In particolare, dovrebbe essere eseguita una RM con mezzo di contrasto (Gadolinio), che però non fu praticato nel nostro caso. Come descritto in letteratura (9, 10), anche nella nostra paziente, dalla RM dell’encefalo si evidenzio’ una chiara asimmetria degli emisferi cerebrali; in partcolare, appare aumentato lo spessore della sostanza bianca omolaterale alle lesioni angiomatose. La prevalenza di emiipertrofia cerebrale, nel campione di 11 pazienti con KTS dello studio esaminato, fu del 18%; nessuno di tali pazienti aveva malformazioni vascolari intracraniche (9). Il tessuto angiomatoso ipertrofico mastoideo non ha impedito la corretta esecuzione dell’intervento. La sua rimozione non ha causato lesioni del nervo facciale e a distanza di sei mesi dall’intervento l’impianto funziona correttamente con conseguente notevole miglioramento del linguaggio.
Bibliografia
1. Klipple M, Trenaunay P. Du naevus variquex osteohypertrophique. Arch Gen Med. 1900;3:641–72
2. Cohen, M. M. (2000), Klippel-Trenaunay syndrome. Am. J. Med. Genet., 93: 171–175.
3. Lamar L. M., Farber G. A., O” Quinn S. E. Klippel-Trenaunay-Weber syndrome. Arch. Dermatol.1965;91:58–58.
4. Sadiq MF, Shuaib W. Klippel-Trenaunay syndrome with intracranial arteriovenous malformation: a rare presentation. Case Rep Radiol. 2014;2014:20216
5. Makiyama Y, Nishimoto H, Fukaya C, Tsubokawa. Massive intracerebral hematoma in a child with Klippel-Trenaunay syndrome. Surg Neurol. 1994 Nov;42(5):392-5; discussion 395-6
6. Fierek O, Laskawi R, Kunze E. Large intraosseous hemangioma of the temporal bone in a child. Ann Otol Rhinol Laryngol. 2004 May;113(5):394-8
7. Oduber CE1, van der Horst CM, Hennekam RC. KlippelTrenaunay syndrome: diagnostic criteria and hypothesis on etiology. Ann Plast Surg. 2008 Feb;60(2):217-23
8. Suver DW1, Perkins J, Manning SC. Klippel-TrenaunayWeber syndrome with labyrinthine bony overgrowth and mixed hearing loss, a case report. Int J Pediatr Otorhinolaryngol. 2004 Aug;68(8):1075-9.
9. Torregrosa A, Martí-Bonmatí L, V Higueras, Poyatos C, Sanchís a. Klippel-Trenaunay syndrome: frequency of cerebral and cerebellar hemihypertrophy on MRI. Neuroradiology. 2000 Jun; 42 (6): 420-3.
10. Hossein Esmailzadeh, Azita Tavassoli, Younes Jahangiri N, Nasibeh Vatankhah, Klippel-Trenaunay-Weber Syndrome with Hemimegalencephaly; Report of a Pediatric Case Iran J Pediatr. 2012 Mar; 22(1): 147–151.
 Fig. 1
Fig. 1
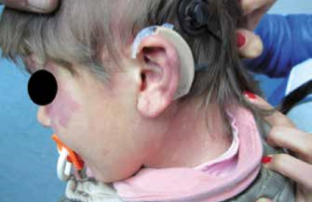 Fig. 2
Fig. 2
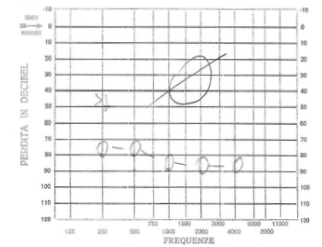 Fig. 3
Fig. 3
 Fig. 4
Fig. 4
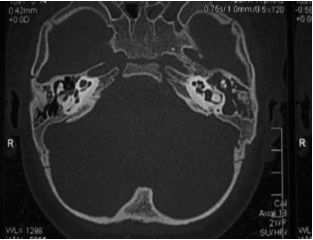 Fig. 5
Fig. 5
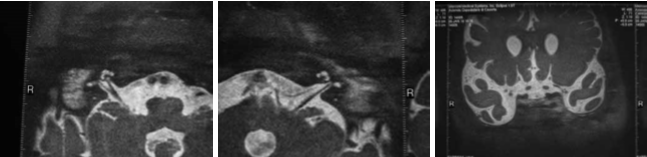 Fig. 6
Fig. 6
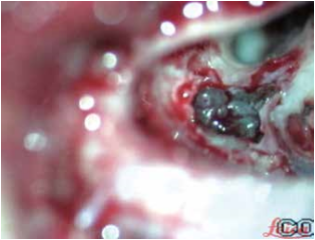 Fig. 7
Fig. 7
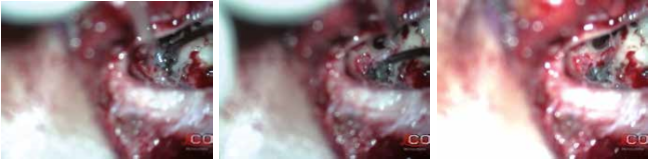 Fig. 8
Fig. 8
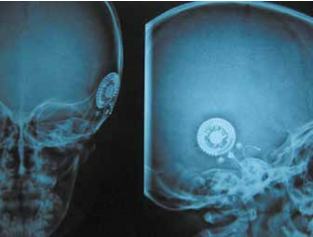 Fig. 9
Fig. 9


